


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
斉藤 淳一; 小林 洋平*; 澁谷 秀雄*
Materials Transactions, 62(10), p.1524 - 1532, 2021/10
被引用回数:5 パーセンタイル:41.35(Materials Science, Multidisciplinary)高速炉の冷却材である液体ナトリウムの新規技術の創出の一環として、液体ナトリウムの濡れ性制御の研究開発を実施しており、本発表はその基礎的研究として、純金属の液体ナトリウムと液体錫の濡れ性を接触角により評価した。また、界面の原子間相互作用を界面クラスターモデルを用いて分子軌道法により理論計算した。その結果、界面の原子間結合力と濡れ性には相関があることが明らかになった。この知見は今後、ナトリウム濡れ性を制御するうえで、重要な情報となる。
斉藤 淳一; 小林 洋平*; 澁谷 秀雄*
日本金属学会誌, 85(3), p.110 - 119, 2021/03
被引用回数:1 パーセンタイル:8.06(Metallurgy & Metallurgical Engineering)Wettability of pure transition metals with liquid sodium or liquid tin has been evaluated using a contact angle droplet method. Titanium, iron, nickel, copper and molybdenum pure metals were selected as specimens in this experiment. All experiments were carried out in high purity argon gas and extremely low moisture to avoid influence of oxygen to liquid metal. Measurement temperature was set just above the melting temperature of each liquid metal. As a result, for both liquid sodium and liquid tin, the measured contact angle changed depending on the atomic number of substrate metals. An electronic structure of the interface between liquid metal and substrate metal was calculated by the molecular orbital method. Simple cluster models of the interface between liquid metal and substrate transition metals were used in this calculation. It was found that the calculation results well express an electronic state of interface. The atomic bonding between liquid metal atom and substrate metal atom changed depending on the kind of substrate metal. Also, the atomic bonding between substrate metal atoms changed similarly. It became clear that there was a reasonable relationship between an atomic bonding ratio and the contact angle. It clearly suggested the atomic bonding affected wettability between liquid metal and substrate metal. The atomic bonding was obtained as one of indications to reveal the wettability by transition metals with liquid metals.
城戸 健太朗; 笠原 健人*; 横川 大輔*; 佐藤 啓文*
Journal of Chemical Physics, 143(1), p.014103_1 - 014103_9, 2015/07
被引用回数:5 パーセンタイル:14.86(Chemistry, Physical)In this study, we reported the development of a new quantum mechanics/molecular mechanics (QM/MM)-type framework to describe chemical processes in solution by combining standard molecular-orbital calculations with a three-dimensional formalism of integral equation theory for molecular liquids (MC-MOZ method). The theoretical procedure is very similar to the 3D-RISM-SCF approach. Since the MC-MOZ method is highly parallelized for computation, the present approach has the potential to be one of the most efficient procedures to treat chemical processes in solution. Benchmark tests to check the validity of this approach were performed for two solute (solute water and formaldehyde) systems and a simple S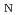 2 reaction (Cl
2 reaction (Cl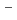 + CH
+ CH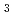 Cl
Cl  ClCH + Cl) in aqueous solution. The results for solute molecular properties and solvation structures obtained by the present approach were in reasonable agreement with those obtained by other hybrid frameworks and experiments. In particular, the results of the proposed approach are in excellent agreements with those of 3D-RISM-SCF.
ClCH + Cl) in aqueous solution. The results for solute molecular properties and solvation structures obtained by the present approach were in reasonable agreement with those obtained by other hybrid frameworks and experiments. In particular, the results of the proposed approach are in excellent agreements with those of 3D-RISM-SCF.
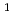 D)+N
D)+N ONO+NO
ONO+NO河合 信之輔*; 藤村 陽*; 梶本 興亜*; 高柳 敏幸
Journal of Chemical Physics, 120(14), p.6430 - 6438, 2004/04
被引用回数:8 パーセンタイル:25.17(Chemistry, Physical)O(D)+NO反応で生成するNO(v=0,1,2)の回転状態の分布を測定した。回転温度はおよそ20000Kであり、分布は位相空間理論で予想されるものに近いことがわかった。この結果は、反応中間体の寿命がそれほど長くはないが、分布はほぼ統計的であることを意味する。しかしながら、回転量子数の大きな場合には、分布は位相空間理論で予想されるよりも早く減衰した。このことを理解するため、分子軌道計算に基づいたポテンシャル曲面を用いて古典軌道計算を行った。その結果、実験で得られた高い回転量子数の分布が反応出口領域のポテンシャルの影響を強く受けることがわかった。
D) insertion into the C-C bond in cyclopropane and ethane黒崎 譲; 高柳 敏幸
Chemical Physics Letters, 355(5-6), p.424 - 430, 2002/04
被引用回数:3 パーセンタイル:8.65(Chemistry, Physical)シクロプロパンのC-C結合に対するO(D)挿入反応の入り口付近における5つの最低一重項ポテンシャルエネルギー面を、CASPT2/cc-pVDZレベルで計算した。その結果、5枚のポテンシャル面の内の最も下にあるものは、入り口付近で引力的であるのに対し、他の4枚は斥力的であることが予測された。比較のため、エタンについて同様の計算を行った結果、5枚のポテンシャル面は入り口付近ですべて斥力的であることが予測された。これらの計算結果は、O(D)とアルカン分子の反応についての最近の実験結果と矛盾しない。
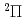 )+N(X
)+N(X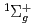 )
)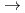 HCN(X
HCN(X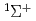 )+N(
)+N(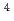 S) reaction
S) reaction和田 晃; 高柳 敏幸
Journal of Chemical Physics, 116(16), p.7065 - 7072, 2002/04
被引用回数:10 パーセンタイル:30.29(Chemistry, Physical)スピン禁制反応 CH(X)+N(X)HCN(X)+N(S) について、量子散乱理論を用いた計算を行った。CH分子を一個の原子とみなすことによって、自由度を3次元に落とした。分子軌道計算を用いて、スピン2重項及び4重項それぞれのポテンシャルエネルギー曲面を作製した。また、スピン軌道相互作用については過去の理論計算を用いた。超球座標を用いた堅密結合方程式を数値的に解いて、総反応確率を計算した。計算された確率は典型的な共鳴構造を示した。得られた確率から反応速度定数を計算し、実験結果と比較したところ、100倍ほど小さな値が得られたが、速度定数はスピン軌道相互作用に大きく依存することがわかった。
高柳 敏幸; 和田 晃
Chemical Physics Letters, 352(1-2), p.91 - 98, 2002/01
被引用回数:42 パーセンタイル:77.42(Chemistry, Physical)ヘリウムを含んだ化合物であるHHeF分子について、多配置参照配置間相互作用レベルの高精度分子軌道計算を行った。計算の結果、直線分子H-He-Fは準安定で、H-He-FH+He+Fの解離に対して0.224eVのエネルギー障壁をもち、H-He-FHe+HFの反応に対しては、0.448eVのエネルギー障壁をもつことがわかった。分子軌道計算を約3000点について行い、グローバルなポテンシャルエネルギー曲面を補間法によって作製した。そのポテンシャル面を使って3次元の時間に依存した波束計算を行ったところ、準安定共鳴状態の寿命は157fsと見積もられた。
H +Cl reaction in water; Ab initio molecular orbital study using a cluster model
+Cl reaction in water; Ab initio molecular orbital study using a cluster model黒崎 譲
Journal of Physical Chemistry A, 105(49), p.11080 - 11087, 2001/12
被引用回数:3 パーセンタイル:9.19(Chemistry, Physical)CH+Cl反応の水中における電荷分離過程を、反応系に4個の水分子を加えたクラスターモデルを用いて非経験的分子軌道法により研究した。その結果、最終生成物である電荷分離錯体(M3)は反応系の解離極限、CH+Cl+4HO、より4.3kcal/molエネルギー的に低く、反応系で生成する錯体(M1)より0.6kcal/mol高いことが予測された。また、気相中の自由エネルギーを求め、それに溶媒和自由エネルギーを加えることによって全自由エネルギーを計算したところ、M3の全自由エネルギーはM1より5.1kcal/mol低いことが予測された。この結果は水中におけるこの反応系の電荷分離過程が自発的であることを強く示唆する。
 molecular orbital theory and path integral molecular dynamics
molecular orbital theory and path integral molecular dynamics志賀 基之; 立川 仁典*; 三浦 伸一*
Journal of Chemical Physics, 115(20), p.9149 - 9159, 2001/11
被引用回数:120 パーセンタイル:95.29(Chemistry, Physical)経路積分分子動力学法を核に対して用い、非経験的分子軌道法を電子に対して適用することによって、核も電子も量子力学的に取り扱うことのできる第一原理計算法の一般論を展開した。この方法は、ボルン-オッペンハイマー近似の枠内で、原子配置と電子構造の両方に依存する物理量を求めることを可能にした。また、応用として、一様電場に対する応答量(双極子モーメントや分極率)の有限温度での定式化を行い、核分布と電子分布が温度や同位体置換効果に対して受ける影響を定量的に計算する方法を提案した。また、水分子の計算を実施し、実験と良い一致が見られることを示した。
D)+NONO+NO reaction高柳 敏幸; 和田 晃
Chemical Physics, 269(1-3), p.37 - 47, 2001/07
被引用回数:14 パーセンタイル:41.27(Chemistry, Physical)O(D)+NONO+NO反応について、量子反応性散乱計算を行った。ポテンシャルエネルギー曲面は、CASPT2レベルの高精度の分子軌道計算を行い、解析関数にフィットして作製した。反応側及び生成側の配向角を固定したモデルを用いることによって、次元を3次元に落とした。この反応では2種類のNO分子が生成する。反応熱はおもに新しく生成するNO分子の振動に分配されるが、もともと存在したNO振動モードにも、ある程度エネルギーが分配されることを見出した。このことはもともと存在したNO結合が、必ずしもスペクテータではないことを示している。
望月 祐志*; 小出 洋; 今村 俊幸; 武宮 博*
Journal of Synchrotron Radiation, 8(Part.2), p.1003 - 1005, 2001/03
アデニンとグアニン、この2つのプリン塩基分子は、DNAを構成する重要な化合物だが、これまでその物性は原子価電子によるものがおもに取り上げられてきた。原子価電子は非局在性が高いため、原子の局所的情報を得るには不向きである。一方、内殻電子にかかわるX線吸収スペクトルは、Xの局在性故に各原子の局所的化学環境が調べられている。この研究では、プリン塩基の窒素K殻スペクトルを、プリン環への化学修飾、水和などを組み合わせた一連の系へのHF-STEX,RASSCF計算によりシミュレーションし、窒素原子K殻吸収端エネルギーがいかにシフトするか系統的に評価する。この計算の遂行には大規模な並列処理にするが、将来さらに大型の計算も可能とするよう、異機種計算機上での分散並列への対応も合わせて進めている。
志賀 基之; 立川 仁典*; 三浦 伸一*
Chemical Physics Letters, 332(3-4), p.396 - 402, 2000/12
被引用回数:73 パーセンタイル:89(Chemistry, Physical)化学結合の組み換えや分子の電子状態・振動状態の動的変化によるエネルギーの授受など、化学反応において競定しうるさまざまな基本的過程において、水素など軽い原子の量子的性質が重要であると考えられている。このような系ではポテンシャル曲面の微妙なふるまいが反応速度に劇的な影響をもたらす。そこで本研究では、電子だけでなく原子核をも第一原理的に量子力学的取り扱いをしたシミュレーションを提案し、あらゆる物質系に応用できる一般的手段としての数値計算法とその応用例を実証した。すなわち、電子状態に対してハートリー・フォック近似を出発点としているLCAO-MO法,原子核に対しては経路積分分子動力学法を用いた計算手法の詳細を示し、水分子の各原子核の量子ゆらぎをその電子状態への影響について計算結果を報告した。
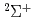 ) with CH, CH, HO and their isotopic variants
) with CH, CH, HO and their isotopic variants梅本 宏信*; 寺田 直樹*; 田中 邦和*; 高柳 敏幸; 黒崎 譲; 横山 啓一
Chemical Physics, 259(1), p.39 - 47, 2000/09
被引用回数:9 パーセンタイル:27.49(Chemistry, Physical)NO分子の第一励起状態Aとアセチレン,エチレン及び水との反応において、水素原子が直接生成することを初めて実験的に確認した。水素原子のドップラー分光の結果からアセチレンと水については1/4のエネルギーが並進運動に分配され、エチレンについては1/7であった。この結果は反応過程で極めて寿命の短い中間体が生成していることを示すものである。反応のメカニズムをさらに詳細に理解するため、ab initio分子軌道法によるポテンシャルエネルギー曲面の計算を行った。
 D)+CH reaction; Does the N(D) atom really insert into CH bonds of alkane molecules?
D)+CH reaction; Does the N(D) atom really insert into CH bonds of alkane molecules?高柳 敏幸; 黒崎 譲; 横山 啓一
International Journal of Quantum Chemistry, 79(3), p.190 - 197, 2000/09
被引用回数:11 パーセンタイル:49.22(Chemistry, Physical)最近、米国の量子化学研究者によってN(D)原子がメタンのCH結合に挿入しないことが報告されたが、本論文はその研究結果に対する反論である。多配置ハートリーフォック計算、さらに大規模な配置間相互作用を考慮した計算によって、N(D)原子がCH結合に挿入してCH NH(A'')を生成することを改めて理論的に示した。さらに興味深いことに二重項第一励起状態のポテンシャル曲面上でも挿入反応が起こることを見いだした。この場合はCHNH(A')分子が生成する。これらの結果は最近われわれが行ったN(D)+Hのポテンシャル曲面の結果とよく似ている。
NH(A'')を生成することを改めて理論的に示した。さらに興味深いことに二重項第一励起状態のポテンシャル曲面上でも挿入反応が起こることを見いだした。この場合はCHNH(A')分子が生成する。これらの結果は最近われわれが行ったN(D)+Hのポテンシャル曲面の結果とよく似ている。
+CHH+CH reaction and its isotopic variants黒崎 譲; 高柳 敏幸
Journal of Chemical Physics, 113(10), p.4060 - 4072, 2000/09
被引用回数:22 パーセンタイル:55.85(Chemistry, Physical)反応H+CHH+CH(1)及びこれを同位体置換した反応、HD+CHH+CHD(2), DH+CHD+CH(3),D+CHD+CHD(4),H+CDH+CHD(5)の反応速度定数を、トンネル補正を加えた変分的遷移状態理論により計算した。その結果、これらの反応に見られる同位体効果はほとんど一次同位体効果によるもので、二次同位体効果及び反応経路(IRC)の曲率の効果は比較的小さいことが明らかとなった。このことは、分子軌道計算からも明らかなように、これらの反応のポテンシャルが「early」であることに起因すると思われる。また、反応1と2の反応速度定数の計算結果は、実験結果とかなり良い一致を示した。
NO(B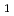 )NO(
)NO(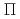 )+H reaction
)+H reaction黒崎 譲*; 高柳 敏幸
Journal of Molecular Structure; THEOCHEM, 507(1-3), p.119 - 126, 2000/07
反応HNO(2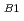 )NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
)NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
H+ClCHCl reaction黒崎 譲*
Journal of Molecular Structure; THEOCHEM, 503(3), p.231 - 240, 2000/05
本研究では、まず気相中における反応CH+ClCHClの機構について理論的に検討した。極限的反応座標(IRC)計算の結果、得られた反応物、遷移状態(TS)、生成物が1つの反応経路上にあることが確認された。反応の活性化エネルギーはPMP4及びB3LYPレベルで、それぞれ36.3,35.9kcal/molと計算された。次に、溶媒中における同反応の機構について検討した。その結果、極性溶媒中では気相中のようなTSが存在しないことが予測された。また、気相中では励起状態であったCHCl +Cl
+Cl が、極性溶媒中では基底状態となることが明らかとなった。誘電率80の極性溶媒中では、CHCl+Clのエネルギー値は反応物(CH+Cl)と比較して11.2kcal/molとなることがB3LYPレベルで計算された。このことは、この反応が気相中よりも極性溶媒中でより起こりやすいことを示唆している。
が、極性溶媒中では基底状態となることが明らかとなった。誘電率80の極性溶媒中では、CHCl+Clのエネルギー値は反応物(CH+Cl)と比較して11.2kcal/molとなることがB3LYPレベルで計算された。このことは、この反応が気相中よりも極性溶媒中でより起こりやすいことを示唆している。
Balucani, N.*; Algia, M.*; Cartechini, L.*; Casavecchia, P.*; Volpi, G. G.*; 佐藤 圭*; 高柳 敏幸; 黒崎 譲*
Journal of the American Chemical Society, 122(18), p.4443 - 4450, 2000/05
被引用回数:77 パーセンタイル:87.95(Chemistry, Multidisciplinary)第一励起状態であるN(D)原子のアセチレンとの反応について、公差分子線と高いレベルの分子軌道計算によって調べた。主たる反応メカニズムはN(D)+CHHCCN+Hであり、窒素と水素原子が交換する。この反応はタイタンの大気化学に非常に重要であることが予想される。これまで大気中のCHを含んだ化合物はほとんどイオン分子反応で生成すると考えられていたが、本研究は中性分子間の反応も重要であることを示す。
S, D, P)+H reactions高柳 敏幸; 黒崎 譲; 横山 啓一
Chemical Physics Letters, 321(1-2), p.106 - 112, 2000/04
被引用回数:24 パーセンタイル:58.73(Chemistry, Physical)多配置参照配置間相互作用の方法を用いた分子軌道法によってN(S, D, P)+Hの反応のポテンシャルエネルギー曲面を計算した。特にC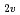 とCoor配置の計算を重点的に行い、2次元のポテンルシャル面を詳細に作製した。その結果、N(D)+H反応については5枚のポテンシャル面のうち、2枚が重要であることを明らかにした。またこれらのポテンシャル面が最低4重項のポテンシャルと交差し、N(S)+Hへの非断熱遷移が起こりうることを見いだした。また計算結果に基づき、N(P)+Hの消光過程のメカニズムについて検討した。
とCoor配置の計算を重点的に行い、2次元のポテンルシャル面を詳細に作製した。その結果、N(D)+H反応については5枚のポテンシャル面のうち、2枚が重要であることを明らかにした。またこれらのポテンシャル面が最低4重項のポテンシャルと交差し、N(S)+Hへの非断熱遷移が起こりうることを見いだした。また計算結果に基づき、N(P)+Hの消光過程のメカニズムについて検討した。
+HCH+H and CD+HCDH+H reactions using variational transition state theory and the multidimensional semiclassical tunneling method黒崎 譲*; 高柳 敏幸
Journal of Chemical Physics, 110(22), p.10830 - 10842, 1999/06
被引用回数:20 パーセンタイル:53.61(Chemistry, Physical)反応CH+HCH+H(I)及びCD+HCDH+H(II)の反応速度における同位体効果について、変分的遷移状態理論及び準古典的多次元トンネリング法を用いて理論的に考察した。まず、反応IとIIのポテンシャル面を量子化学的手法により計算した。次に、得られたポテンシャル面を用いて、多次元トンネリングを準古典的に考察した変分的遷移状態理論により反応速度定数を求めた。実験的には、5Kの固体パラ水素中で、反応IIの方が反応Iより反応速度が速いことが報告されている。ここでの計算の結果、理論的にも反応IIの方が反応Iよりも5Kで反応速度が速いことが予測され、実験結果を定性的に説明することができた。
